蛋白质三维结构与 AlphaFold2 之间不得不说的故事-1
2021年7月23日,笔者的朋友圈被AlphaFold2 的新闻刷屏了。不得不说,Google再一次通过自身强大的计算资源和技术优势,让世界瞠目结舌。本文意在向各位读者介绍一下蛋白质三维结构与 AlphaFold2 的一些故事。
1.罗马不是一天建立的
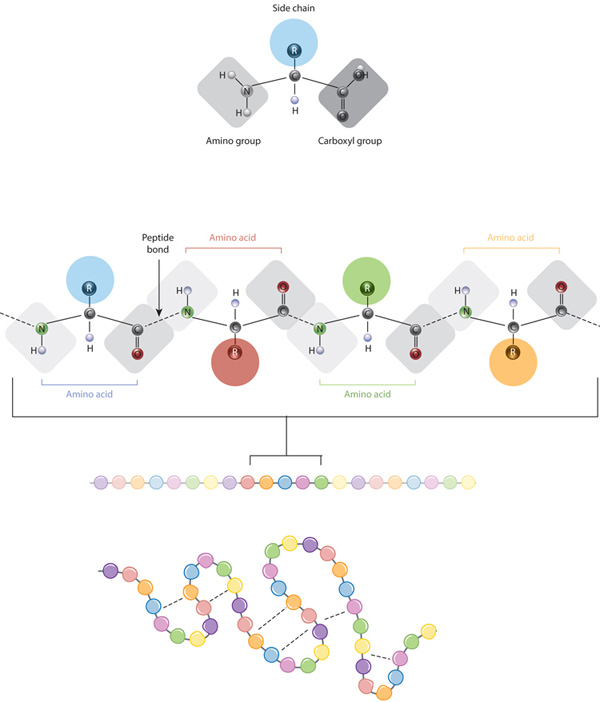
如果说新中国科学家离诺贝尔奖最近的一次,大概就应该是"人工合成牛胰岛素"。虽然其中的成年旧事不是本文的核心,但是这个重大的科研成果还是对生命科学的研究起到了重大推动作用。那就是人类首次得出了一个重要的结论:“蛋白质的三级结构只取决于其氨基酸序列”。这也就是说,DNA序列本身记录的信息,不仅仅蕴含着氨基酸序列信息,更隐含了蛋白质这个生命活动的载体的全部信息。
这就引出了另外一个问题,即“氨基酸是如何决定了蛋白质三维结构的?”。这一问题,毫不夸张地说,如果,沃森和克拉克(诺奖获得者)解决了DNA双螺旋结构,解释了遗传学第一重迷雾;马特伊与尼伦伯格(诺奖获得者)破解了密码子表,解释了遗传学的第二层迷雾;那么谁能解释清楚蛋白质是如何由肽链折叠形成的,也应该是诺贝尔奖级别的科研发现了。
2. 深度学习参与前的努力
2.1 实验方法获得蛋白结构信息
从整体而言,在目前比较靠谱的蛋白结构解析有两种方式: 一是实验法,常见的技术有X晶体衍射、核磁共振法,冷冻电镜等。这类方法得到的结果信息准确,可验证性高。早年间甚至有解决了一个蛋白结构(比如膜蛋白)就可以博士毕业后直接博导的说法(现在显然是不可能了)。但是实验方式存在一个较大的问题,那就是过于耗时耗力。早年曾经听带我的师兄曾经说过一句非常总结性的话:“蛋白结晶是一门艺术”。
另一方面,对于核磁共振法而言,对蛋白本身的解析能力其实是有限的,所以应用上,更适合解决部分肽段的结构,而不适合于解析整个蛋白的结构。目前来看,冷冻电镜可以算得上一个实验方法上的革命,但是其目前也有一定的限制。后面会找机会详细说明。另一个方式,就是通过计算预测。这也是彻底解决蛋白质三维结构问题的方法。
2.2 PDB数据库
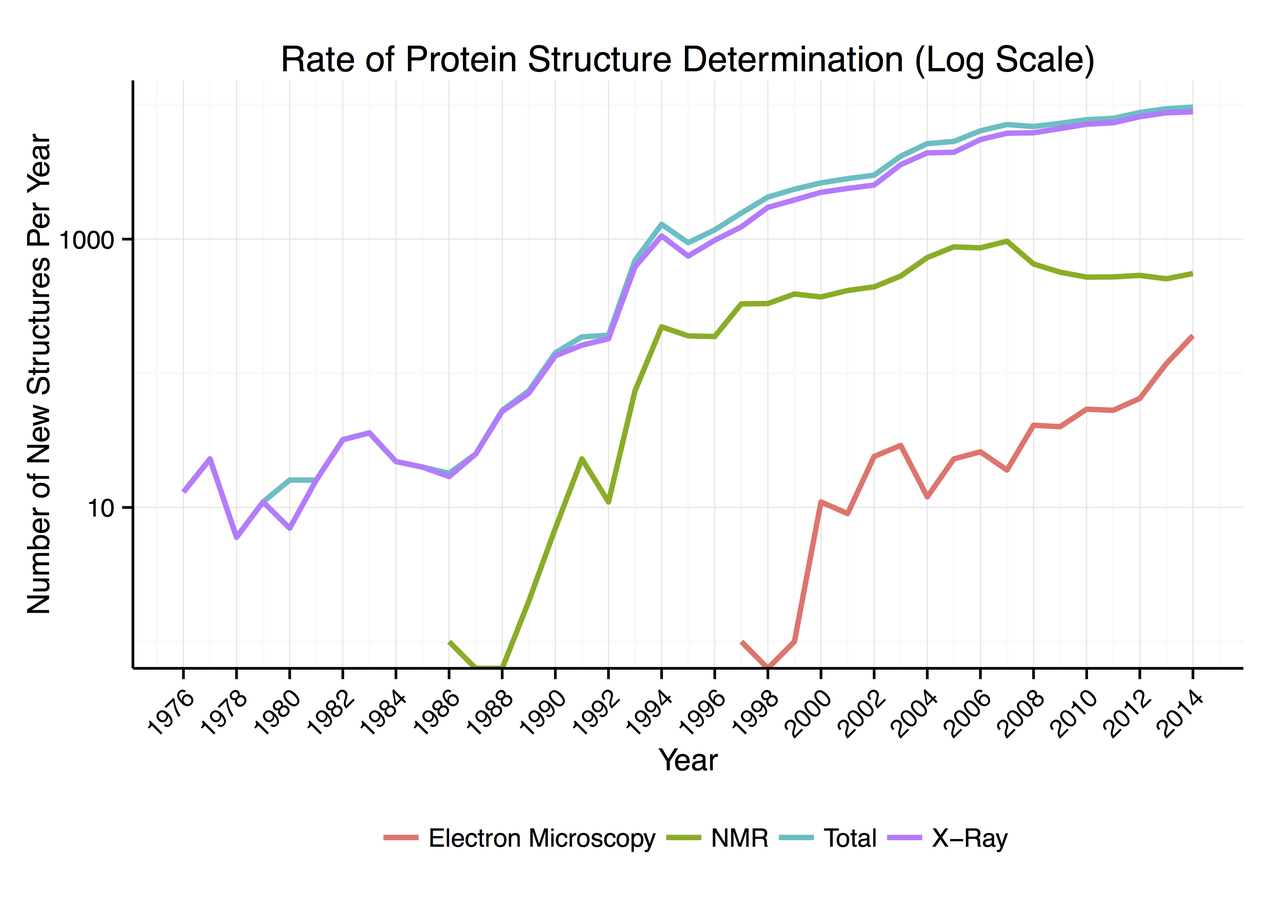
蛋白质数据库(Protein Data Bank,简称PDB)是一个专门收录蛋白质及核酸的三维结构资料的数据库。目前是世界范围内较为权威的蛋白三维结构数据库,也是本次AlphaFold2 主要的训练数据集来源。其记录的数据,最主要的来源为X晶体衍射信息,因此数据质量具有较高的可靠性。自2007年开始,该数据库的数据增长量开始放缓…
3.计算方法解决蛋白三维结构问题
3.1 三种主流思路
目前,解决蛋白三维结构的思路主要有三个:
- 根据蛋白之间同源性对结构进行预测。
- 蛋白结构Threading,即根据部分解析出来的核心区域(Template),结合计算对蛋白整体结构进行预测。
- 从头预测法(ab initio),其约束条件即能态最低。其基本原理属于物理学范畴。
从目前应用层面看,完全重新预测显然是计算成本最高的方法。第一种通过蛋白同源性进行进行比较的方式,过度依赖已知的数据,因此在应用层面存在较大的限制。所以目前主流的算法,较多集中于第二种,即依赖于已知核心区域,对蛋白整体结构进行预测。对于AlphaFold2而言,其方法其实还是用到了多种方法的,其相关文章中指出:
The methodology we have taken in designing AlphaFold is a combination of the bioinformatic and physical approaches: we use a physical and geometric inductive bias to build components that learn from PDB data with minimal imposition of handcrafted features (e.g. AlphaFold builds hydrogen bonds effectively without a hydrogen bond score function). This results in a network that learns far more efficiently from the limited data in the PDB but is able to cope with the complexity and variety of structural data.
3.2 CASP 竞赛
本期最后,再介绍一下CASP竞赛。对于蛋白质三维结构预测这样的殿堂级问题,全世界范围的"通(ge)力(zi)合(wei)作(zhan)"肯定是要有的。因此,为了防止哪个国家暗地里憋大招(例如我们的邻居朝鲜),因此有了CASP竞赛,这是其官方网址。
其竞赛美两年举行一次,自1994年开始,距今已经成功举办了14次了,其原理也很简单,给出一组蛋白或肽段的fasta文件,通过各个实验室的服务器进行计算,与目标蛋白或肽段真实结构进行比较,预测结果与真实结果最为接近的代表队胜出。也就是在这个竞赛中,AlphaFold2 才大放异彩。