变异记录文件格式 vcf
vcf文件做记录个体或群体突变的文件格式,在生物信息学应用中举足轻重。主流的生物信息分析软件,在处理变异信息时,也基本上需要考虑支持解析或输出vcf格式的文件。本文在介绍vcf文件格式的基本格式的同时,对vcf文件记录的细节进行描述。希望对广大开发者和生物信息学从业人员起到帮助。
1.vcf文件概述
vcf文件格式是变异结果存储的标准格式,一般多用于单核苷酸变异(SNV)或小片段的插入缺失(indels)的结果记录。除此之外,vcf文件也可以存储其他变异形式,比如CNV(拷贝数变异)、SV(结构变异)等,但目前难以形成主流。基因组结构类变异,目前相对较多的依然是bed或bedpe文件,后面会陆续为各位进行介绍。
SNV 是基因组上单个位置的替换。|比如,在参考基因组上记录为 A ,通过检测,某个体由于个体差异或突变,在相同位置变异为C。Indel是指插入或缺失,例如在参考基因组上,某位置为ATCCA,在个体基因组上为ACA(中间位置的TC缺失),则记为A--CA,该位置为deletion。同理若个体基因组存在插入(insertion)情况,与deleltion一样也可以进行记录。insertion和deletion合称为indel。
vcf文件主要有三种模式:
- 第一种为仅有位点信息,即对变异发生的位置和变异本身
- 第二种为个体变异记录的是某个个体或个体组织的突变情况
- 第三种为群体变异检测信息,记录的为突变在群体或家系中发生情况。
这三种类型文件虽然在记录内容上有所差别,但是都遵循vcf的基本规则。下图为vcf文件实例:
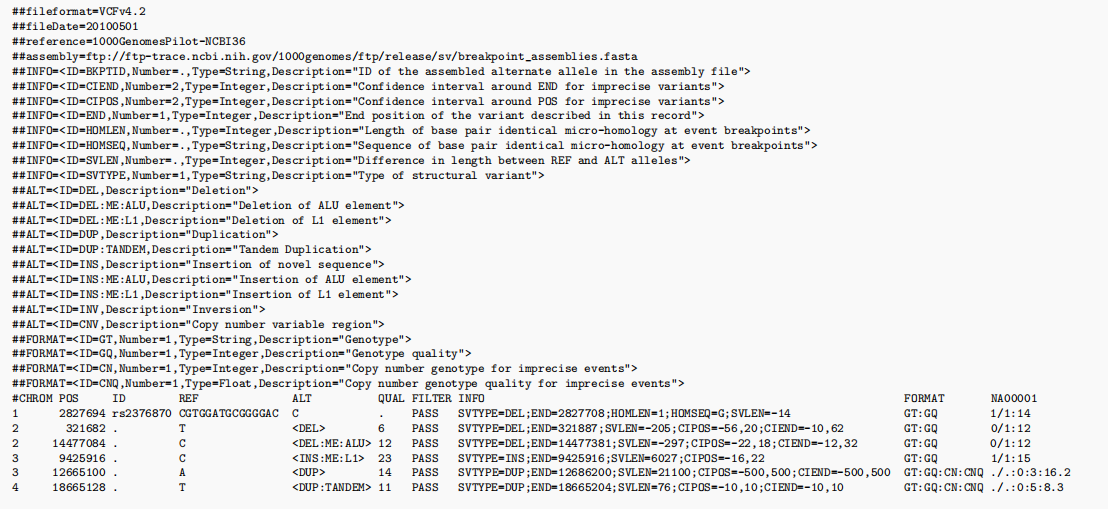
注意: 从上述说明中可以看出,单核苷酸的记录其实相对容易,但是对indel变异而言,由于插入缺失片段的长度不定,其位置并非固定的,因此在样本间的记录难以统一。
2. vcf meta 信息部分
vcf文件头部主要记录文件原信息(meta information),每行以##开始,主要记录内容包括下列内容
| 记录内容 | 说明 |
|---|---|
| 文件格式(fileformat) | 记录文件格式;主要记录本文件所采用的vcf格式版本 |
| 信息区域(INFO)描述 | 该部分记录的是作为INFO区的基本描述,由于在vcf记录中,对INFO内容采用的是缩写,因此在此部分进行会对其含义以及数据类型进行说明。 |
| 过滤方式(FILTER) | 该部分主要记录vcf文件经过生成过程中经过哪些过滤程序处理。记录包括命令行及对应参数 |
| 个体区域格式信息(FORMAT) | 对于每个样本,vcf正文部分会记录多种信息,例如变异对应深度,该部分记录个体信息中每个字段含义及类型 |
| 突变类型(ALT) | 标识出现在正文信息中突变类型,例如,Insertion、Deletion、Duplication 等 |
| 基因组信息(assembly) | 标识对应的基因组版本 |
| Contig信息 | 由于同一物种肯定存在多个基因组版本(如人类目前常用的有hg19和hg38两个版本),因此可能存在染色体名称相同,但内容不同的情况。因此该部分记录主要用于记录每个染色体的相信信息 |
| 样本基因组信息 | vcf理论上支持不同样本比对于不同基因组的,该部分记录样本和基因组的对应关系(通常较少使用) |
| 遗传谱系信息( Pedigree) | 记录样本间的亲缘关系 |
3. vcf 列名称及正文
vcf文件meta信息记录结束之后,下一行就是列名称,列名称以#作为起始,主要包括以下几列:
- CHROM:表示该列为变异所在的染色体位置。
- POS:表示变异起始坐标,该部分需要注意的是对于结构变异,如Indel,其坐标记录方式不唯一。
- ID:表示变异ID,一般常用的有rs编号或vep编号等。用户也可以根据具体问题自行设定编号,空缺时记为“
.”。 - REF:表示位点在参考基因组上的记录。
- ALT:表示位点可能出现的变异情况。
- QUAL:以Phred(即-log10)格式表示变异可靠性。通过不同变异检测软件得到的结果中,该项记录一般无可比性。
- FILTER:表示过滤方式该部分用户也可以自定义,对于不同过滤标准可以进行不同的命名;但是对于通过过滤标准的,统一记为
PASS。需要注意的是,对于一般的vcf处理软件来说,FILER只标记某个变异是否通过过滤条件。而不是删除记录。 - INFO:该部分记录的变异的评价性指标以及注释信息,例如
AC表示的等因突变的数量。该部分在不同的变异检测软件给出的结果也是不同的,大多数基础统计可以通过bcftools软件来进行追加。
上述八列信息是vcf中必然含有的部分,对于有样本信息,无论是样本还是单样本,都会有第9列 FORMAT列及后续样本信息列:
FORMAT:个体区域格式信息记录样本变异的记录格式。由于vcf文件样本记录中除了变异本身是否发生之外,还会记录针对变异的其他信息,例如对应位点的深度(AD)、基因型质量(GQ)等。FORMAT字段规定了这些信息的基本格式。
个体信息: 个体信息是记录某个样本的具体变异情况,其包含的基本信息格式标准需要符合记录的FORMAT格式。特别需要注意的是基因型GT信息,如果是没有经过phased数据,以
/分隔两个等位位点;如果经过phased则以|分隔。其余信息,均会在FORMAT字段有详细说明。
3. vcf记录标准问题
虽然vcf可以记录和表述突变具体情况,但是实践中由于不同变异检测工具对变异判定的区别,会造成对于同一个变异信息存在不同纪录的可能性(尤其是Indels类型记录)。因此,对于vcf文件而言,除了要求记录的信息全面这个最基本的要求之外,还需要做到两个标准。
3.1 简约性(Parsimony)
简约性是指,用尽可能少的核苷酸表示变体,而不会将任何等位基因的长度减少到0。其规定主要针对多核苷酸多样性而言(简称为MNP,指连续多处核苷酸发生突变)。下图列举了4种例子:
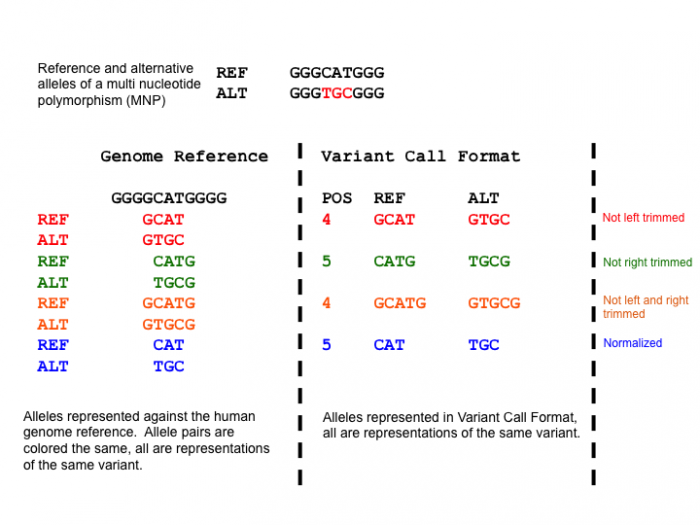
这四种颜色(红、绿、橙、蓝)的对于表示同一突变,其实是等效的。但是前三种记录均不是最简方法。因此,只有第四种蓝色的记录方式,符合简约性规范。
3.2 左对齐(Left alignment)
左对齐主要针对Indel类型的变异。其是指,当且仅当不再可能将其位置向左移动,同时保持其所有等位位点的长度不变时,则变异已左对齐。例如下图:
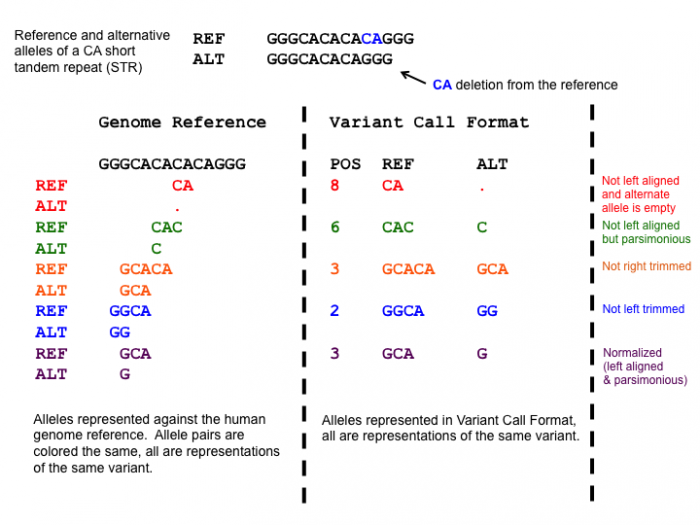
图中给出样本相对对于参考的基因组缺失“CA”碱基的5种记录方式,符合左对齐且简约的,只有最后一种(即紫色)对应的记录方式。
3.3 为什么要遵循上述原则
上述两个原则,其实并不是vcf文件格式规定的,但是相对而言比较重要。尤其对于变异的后期注释、挖掘和使用中,如果记录不满足上述标准,是很难对样本间进行合并或解读。
因此,一般拿到不明来源的vcf文件,比较良好的习惯就是对数据进行标准化处理,再进行其他操作。一般而言,GATK和bcftools都提供了相应的功能,用户可以方便实现。
4.结构变异
vcf文件设计之处,对于结构变异的检测还不想目前这么百花齐放。因此虽然在vcf格式设计上考虑了结构变异的问题,但是相对而言记录比较粗糙。目前各类检测软件虽然基本上都保留了将结构变异输出成vcf的功能。
在vcf中结构变异以以下形式展现
| REF | ALT | 含义 |
|---|---|---|
| s | t[p[ | 片段p在以从右侧开始的顺序,从t所在位置取代了s |
| s | t]p] | 片段 p 反转后(从左侧),从t所在位置取代s |
| s | ]p]t | 片段p在以从右侧开始的顺序,在t之前的位置取代s |
| s | [p[t | 片段 p 反转后(从左侧),从t之前位置取代s |
例如:

由上面的例子可以看出,采用vcf记录结构变异实在不是一个好主意。相比之下,采用bed文件就更加清晰明了。在后面的内容中会向读者陆续介绍。
4. 小结
vcf作为最通用的变异记录文件格式,其格式的表达内容非常丰富,经过压缩后可以快速查询和定位。同时,基于该格式的应用种类相当多,因此是生物信息学重要的文件格式标准之一。
除了格式要求的明规则之外,使用vcf的时候还应该遵守"潜规则",目前主流变异检测软件对规范化的要求明显有所提高,因此最好在使用之前,进行一系列校正。
最后,vcf文件也支持表达结构变异,但是其人工阅读难度和可解析度其实都不太尽如人意。主流的注释工具更多的支持bed或其他格式的结构突变结果。因此,奉劝大家谨慎使用vcf记录结构变异。