生物信息学数据格式
生物信息学作为一门以数据为核心的学科,最为重要的问题,便是将数据按一定的格式进行存储。因此生物信息学最重要的基础,就是正确认识数据文件的结构和格式。因此,本系列从一个完整生物信息分析流的角度,对分析过程中涉及到的文件格式进行梳理。
1. 文件压缩
由于生物信息本身的数据特性,因此,一般最基础的数据文件格式其实都是平面文件,也就是由ascii码编码的文档文件。由这种形式存储的文件,易于编辑和解析,但其占用的存储空间则非常大。以一个原始fastq文件为例,压缩前后数据,为未压缩数据占用存储空间基本上在10分之一左右。虽然在存储空间上有所节约,但是需要说明的一点就是,在某些数据处理过程中,隐含地是需要对文件进行解压操作(例如通过 JellyFish 对k-mer进行统计),因此会造成计算资源消耗。因此对于这部分程序而言,需要通过zcat、gunzip等解压才可能正常运行。
文件压缩,也是一些生物信息操作的必要操作。比如对变异检测结果vcf文件而言,通过基因组位置进行查询是最基本的操作,目前大部分程序都是基于htslib套装中的tabix进行索引操作,进而加速查询过程。htslib可以说是测序相关生物信息领域应用最广的工具之一了(在此膜拜李恒大神及其团队!)。
2. 测序数据 fastq 格式文件
fastq格式是测序下机数据处理的基础格式,也是一般测序公司提供的测序原始数据形式。其记录的就是测序得到的序列以及对应碱基的质量。碱基信息即四种核苷酸(A、T、C、G)。 由于测序过程是生物化学和物理反应(例如Illumina公司的二代测序平台是基于桥式PCR和光学检测手段),过程中不可避免地会产生一定错误。例如光学信号模糊造成的错误识别。因此对于检测结果,需要有一个可度量的评价。碱基质量值(quality score,Q-score),就是反应碱基识别出错概率,Q=-10*lgP,其中P为碱基识别错误概率。 另外,对于序列的识别信息,也需要进一步编码用以方便后续处理中使用,所以fastq文件一共分为4行。是的,虽然整体记录的信息一共三条,但是fastq格式对每个测序序列用4行信息加以记录,示例如下:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
第一行规定以@ 作为开始,记录序列编号。
第二行为测序序列信息,也是最直接的结果。
第三行为+,该行最为特殊,目前为标准的预留行,不记录任何信息。
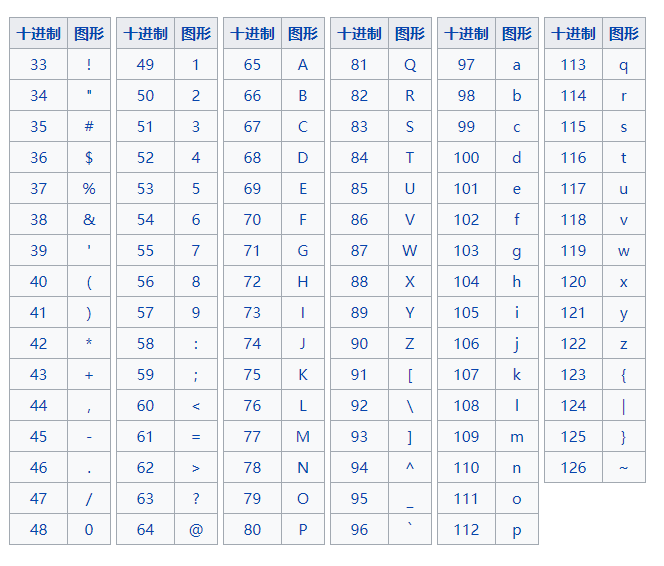
第四行为每个碱基对应的ascii码,用以表征每个碱基的质量。每个字符对应的数值可以通过下面的ascii码值表进行查询。
3. SRA 文件
NCBI作为全球最大的测序数据持有单位,其数据增长量是相当恐怖的。尤其是其作为国际主要期刊公认的原始数据发布平台,其对数据的上传和下载都有较强的需求。在这种压力下,通用的压缩形式就显得略不够看了,因此其提出了一种新的归档方案——SRA (Short Read Archive),用以保存序列。当然,随着技术的进步,尤其是三代测序技术的成熟,SRA 已经不是数据上传至NCBI的唯一选择了。
SRA文件作为一种归档协议下的产物,其天然的不太适合直接进行分析,如果使用其作为分析输入,绝大多数情况下都需要先通过sratools 套件,对数据格式进行转化,生成fastq或bam文件进行下一步分析。sratools作为核心工具,不仅有sra文件处理功能,也兼顾了数据质量控制、sra搜索等诸多功能。除此之外,目前对SRA已经全面拥抱共有云存储环境了(指且仅指 google cloud 和 aws cloud),因此理论上用起来会更爽了。
为什么理论上呢?这个就牵扯到了sra转化成可用的文件格式(fastq或bam)的过程了,不同于通用的压缩算法,其本质是根据基因序列的特别,自己搞了一套编码方式,从而实现了压缩算法。因此,下载到的SRA文件一般都会非常小,而转化过程就非常缓慢…这就是本文第一部分提到,压缩文件的使用,本质就是“以时间换空间”。
4. 作为序列记录的 SAM/BAM 文件格式
sam 文件是 Sequence Alignment/Map 格式的简称,从名字就可以看出,该文件格式设计初衷就是为了记录比对结果的。
但是由于其在记录的比对结果的同时,其实也记录了序列本身,因此也可以作为测序数据的存储格式。上面提到过,sam文件是一种平面文件,也就是没有经过压缩,因此一般占用的存储空间相对较大。因此实际工作中,一般不以sam文件格式作为存储形式,而是采用其压缩格式bam形式。
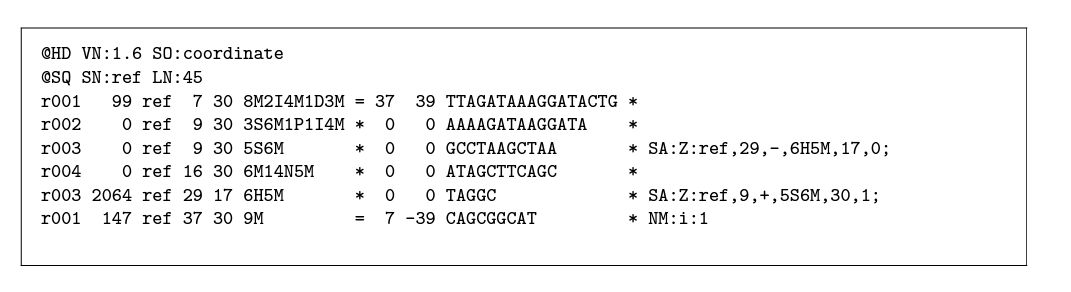
bam 文件是通过 bgzip压缩过的sam文件。因此二者记录的信息本质是一样的。bgzip 对文件的压缩,其实是使用的gzip的方式,其结果使文件被压缩成了一系列的'BGZF block'单元,默认情况下,每个单元大小不超过64K。除了节省存储空间之外,另一个好处就是可以通过建立索引加速查询。在下一篇,将具体为各位介绍bam文件的具体细节,以及相关索引的技术。
三代测序技术中,bam格式和fastq格式是默认测序数据存储的形式之一。早年间的RSII平台,曾经因如果h5存储格式,但是目前不在新平台使用了。
5. FASTA 文件
最后来到了生物工作者基本上都熟悉的fasta格式了,该格式常用于记录参考基因组、蛋白序列、转录本序列等生物大分子序列。这类序列的一个共同点,就是序列相对而言可信度较高。因此,fasta文件中不记录质量值信息,仅记录序列本身和序列编号。例如下面:
>Seq1
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
>Seq2
TGTGGGAGAGGAACATGGGCTCAGGACAGCGGGTGTCAGCTTGCCTGACCCCCATGTCGCCTCTGTAG
该示例中有两条序列,序列编号以行首 > 符号作为开头,占一行,下面记录的则是序列本身,fasta文件支持换行操作,因此并不像fastq文件那样,行数是4的整数倍。
[!WARNING|style:flat] 其实官方规定上,
fastq也支持换行,但是目前主流的处理工具中基本上没有对该特性进行支持,所以事实标准fastq文件的行数是4的整数倍。
除此之外,fasta 文件的也是多重序列比对(MSA multiple sequences alignment)的输出格式之一。但是一般而言,fasta文件的产出量要远远小于fastq 的文件量,因此除了传输过程以外,一般很少对fasta文件进行压缩。因此针对 fasta 的构建索引(例如通过bwa index 构建基因组索引)操作相对来说都比较慢。
6. 小结
本文为大家介绍了基础的序列存储格式,尤其是针对测序仪产生的格式进行了叙述,下一篇会为大家介绍比对结果文件存储格式的一些基础知识。尤其会介绍bam文件的一些信息内容和基础解读套路。希望大家持续关注 :D