DNA测序也有批次效应?
对于测序结果而言,基因型分型数据结果一般而言是最为稳定的内容。从原理上说,一个个体的胚系基因型在出生以来就应该是确定的(免疫细胞除外),因此如果测序是针对DNA的,那么结果一般是确定的。但是事实上,在变异检测方面,不同测序中心、不同年份的数据间确实是存在批次效应的。这种批次效应最直接的体现,就是对结果进行PCA分析时,同一人群中抽样子集存在明显的分离情况。(文献参考:Identifying and mitigating batch effects in whole genome sequencing data 中详细内容)
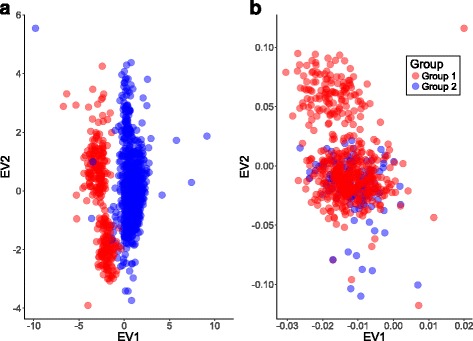
虽然导致变异检测出现批次效应的根本原因尚不明确,但是其直接原因,其实是变异检测过程中存在的错误导致的。目前相对较大型的梯队研究,很难做到 HapMap 或 1000Genome 这种准确性水平——从不同测序分析流程,不同技术手段对大样本量的数据进行分析。因此,在很多情况下,分子遗传学研究人员,不得不与错误突变信息打交道。
批次效应的影响
DNA变异检测的批次效应,最重要的影响就是 : 大规模梯队分析的统计中,统计效力的下降。当分析选用混合线性模型时尤甚。这是因为,对于基于SNP作为分析输入的情况下,数据集的PCA 结果一般反应样本种族特征(这也是微基因这类提供种族溯源分析的公司的基础原理)。当使用混合线性模型进行关联分析的时候,一般会将人口分层结果作为协变量进行分析。而实际上,这部分结果很大可能存在着难以察觉的批次效应。
如何解决批次效应
既然DNA测序数据中批次效应确实存在,那么应该如何规避其带来的不确定性呢?
1. 数据处理流程尽量一致
对于人群梯队研究而言,首先面对的问题就是数据处理问题。就一个测序相关公司的算力来看,大项目的周期显然是很长。在此期间,分析流程的变化是在所难免的。这就需要对流程有比较明确的版本控制和回溯功能。笔者从业多年以来,基本可以确定,对于大型测序公司而言,能做到每个分析项目流程及对应参数都可找回的…嗯…你懂的。
2. 分析流程多样化
DNA比对软件这部分,其实由于李恒大神及其团队的不断努力,俨然已经短序列比对的金标准了。但是变异检测部分,事实证明,还是存在很大空间的。对于常规分析而言,GATK 算是一个不错的选择,但是比如 Freebayes 、Samtools 等工具在检测上也是可以考虑的选项。不过需要注意的是,对于覆盖深度区域,所有目前主流的变异检测工具表现均良好,但对于低深度区域,几个软件之间一致性可能仅为90~80% (文献参考 : Best practices for variant calling in clinical sequencing 中引用数据)。因此,如果对结果要求较高的分析项目,尽量采用多种变异检测工具进行多种分析,是比较好的选择。正如该文章里所指出的:在全基因组范围内,即便是这样的一致性,也能造成不同处理软件结果之间存在几千个不一致的位点。因此,对于负责任的大型项目而言,采用不同种类的变异检测工具是最常用的方法。
3. 变异过滤
传统意义上而言,变异过滤主要是指变异的一系列硬性标准,比如支持reads 数等指标。但是对于不同批次的样本而言,由于测序实际情况不同,采用相同的标准肯定会造成,依据该标准的结果存在批次效应。而GATK 目前最佳实践中推荐的VQSR 过滤标准,其实对该问题也无能为力(其原理是对检测的raw snv 的一系列指标通过高斯混合模型进行训练,再与真集进行比较,获得一个软性标准,进行变异的平分)。
但是随着机器学习的不断进步,目前也有越来越多的基于机器学习的算法,应用到变异过滤领域,比如GATK4 的实验性工具,CNNVariantTrain 、VariFAST 等,都是应用机器学习算法,对变异进行评价的实例。后面我们也会在 www.achelous.org 的生信分析实例中加入相关软件的评测内容。