单细胞免疫组库分析:
编者按:免疫组库的目的是通过可变区的基因测序,获得个体免疫系统整体信息的重要方式,同时也是单细胞测序技术的重要应用之一。此篇从基础角度讲述了如何通过R语言中相关包对数据进行初步的整合和基础分析。在此感谢生信技能树的单细胞天地的本文作者 Kinesin。
1. 10X V(D)J测序简介
研究免疫的朋友一定对V(D)J基因重排表达TCR/BCR的过程了然于心,作为免疫学外行的我就不在此赘述了,我们把重点放在10X V(D)J测序的原理和优势上。
1.1 10X V(D)J测序原理
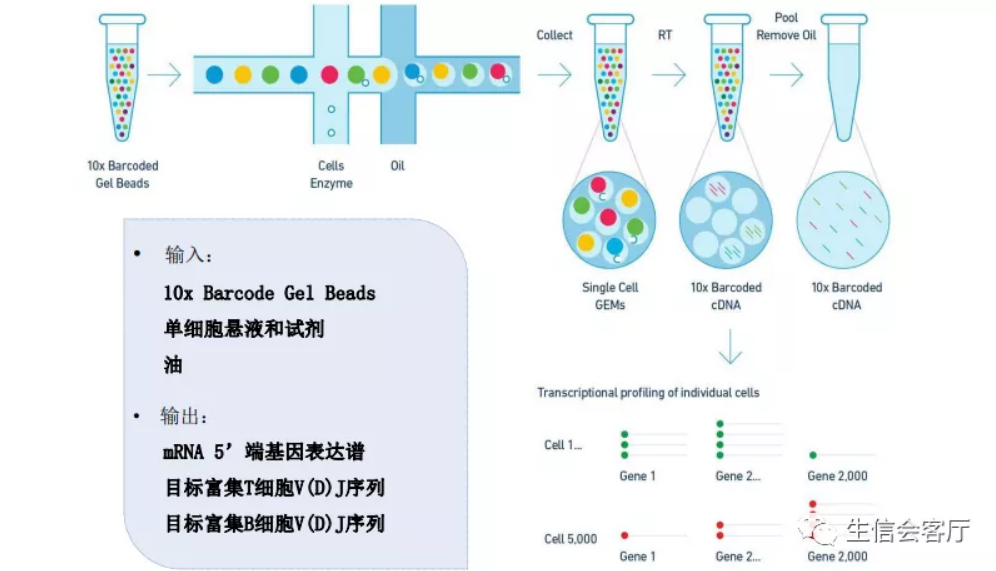
10X V(D)J测序与其转录组测序建库流程大部分相同,都要经历微流控构建单细胞油包水反应体系,mRNA逆转录为cDNA并扩增的过程。与常规10X单细胞转录组测序把barcode和UMI序列放在3'端不同,V(D)J测序要把barcode和UMI序列放在5'端。
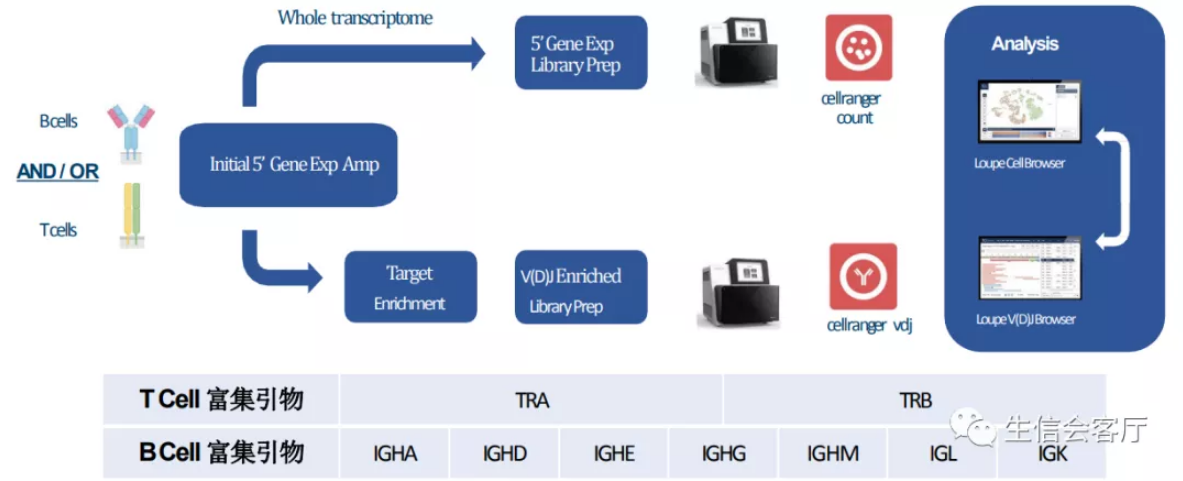
逆转录的cDNA经过扩增后可以一分为二,一部分做5'端scRNA测序,另一部分用富集引物扩增V(D)J序列测序。
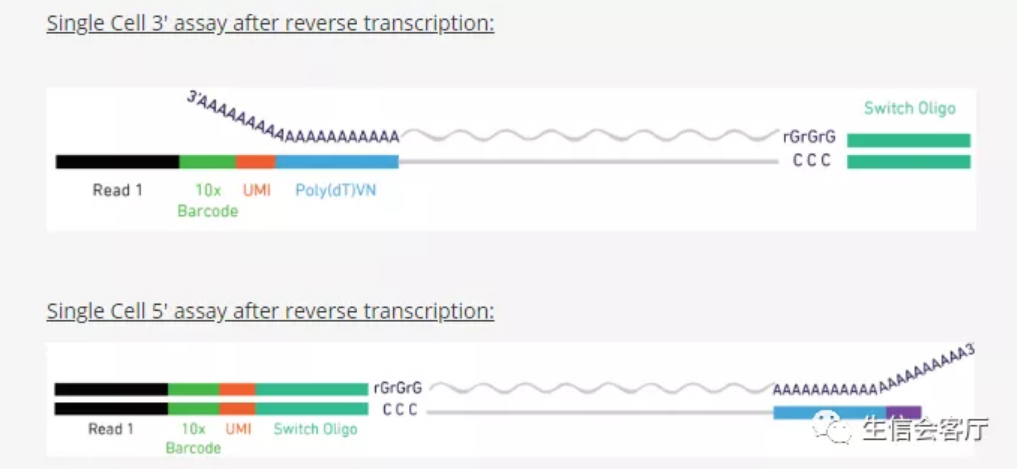
人的V(D)J序列长几百至一千多个碱基,二代测序怎么得到全长序列呢?大家先看看下面的V(D)J测序文库构建原理图:
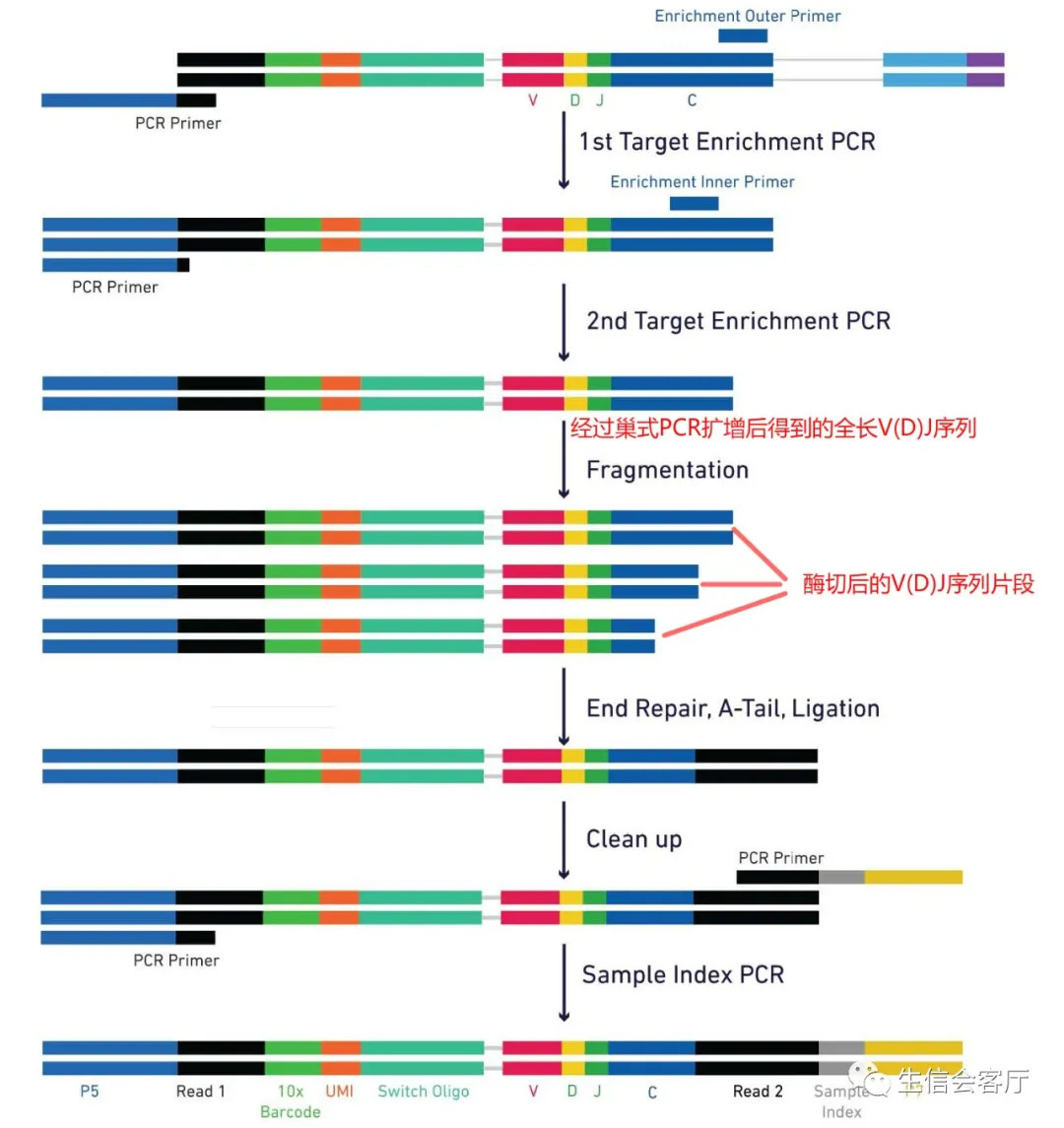
请注意上图中的红色文字,扩增得到的全长V(D)J序列会被酶切为长短不一的片段。因为每条V(D)J序列都有很多个PCR扩增的拷贝,并且这些拷贝有相同的UMI标签,所以每条V(D)J序列都会得到一组测序reads(通过UMI确定同一来源),如下图所示:
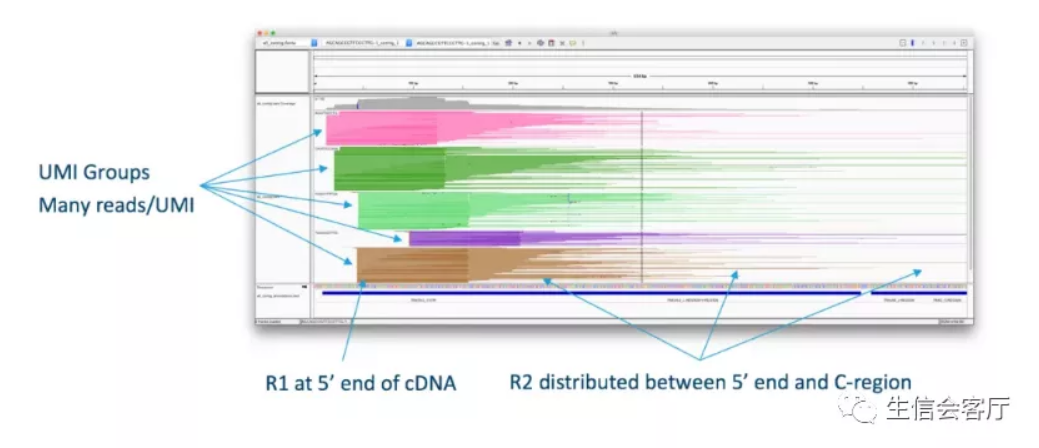
Read1基本固定检测V(D)J序列5‘端150bp的序列,reads2则是随机覆盖V(D)J序列的各个位置(随机打断位置150bp的序列)。只要有足够的数据量,最终测序数据将覆盖全部的V(D)J序列。
1.2 10X V(D)J测序优势

2. scRepertoire简介
scRepertoire由华盛顿大学的Nick Borcherding博士开发,是一款针对10X V(D)J数据的免疫组库分析工具。它直接导入10X cellranger的输出结果,具有丰富的分析项目和美观的可视化结果。 scRepertoire目前发布在github,正式版只支持R4.0,R3.6版本只能使用开发版。开发版相比正式版应该有一些功能上的差异,我运行官方教程时发现有些函数开发版不支持。
library(devtools)
#正式版,需要4.0版R语言支持
install_github("ncborcherding/scRepertoire")
#开发版,适用4.0版以下的R语言
install_github("ncborcherding/scRepertoire@dev")
3. V(D)J数据分析
导入文件说明
scRepertoire需要导入后缀为contig_annotations.csv的CellRanger输出文件,要求barcode没有前缀且以列表的方式传递给scRepertoire。本教程使用scRepertoire包自带的VDJ数据,可通过data("contig_list")加载。
library(Seurat)
library(scRepertoire)
library(tidyverse)
library(patchwork)
rm(list=ls())
dir.create("VDJ")
#加载scRepertoire自带的contig_annotations文件
data("contig_list")
查看contig_list[[1]]的前5行,可以发现这是标准的contig注释数据
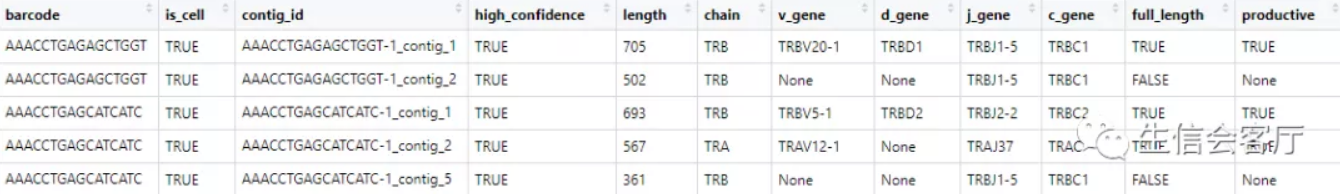
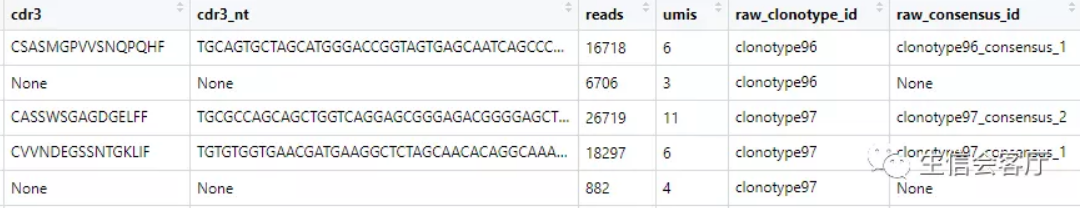
cellranger输出的文件中,没有细胞barcode与clonotype直接对应的数据,因此不能直接整合到seurat中。scRepertoire分析的第一步是把上面的文件转换为barcode与clonotype对应的数据,它通过combineTCR函数实现。
combined <- combineTCR(contig_list,
samples = c("PY", "PY", "PX", "PX", "PZ","PZ"),
ID = c("P", "T", "P", "T", "P", "T"),
cells ="T-AB")
#参数说明:
#samples:用于指定样本名称的参数,因为contig_list包含了6个样本的VDJ数据,所以这里传递了一个6个字符元素的向量;
#ID:用于指定样本名称之外的其他分类信息,例如分组信息;
#cells:指定VDJ类型,"T-AB"代表TCR的α和β链配对,"T-GD"代表γ和δ配对;
#还有removeNA, removeMulti, filterMulti都选择了默认值,有兴趣了解的可以查看帮助文档
输出结果combined依然是一个列表,我们看看combined[[1]]的前5行
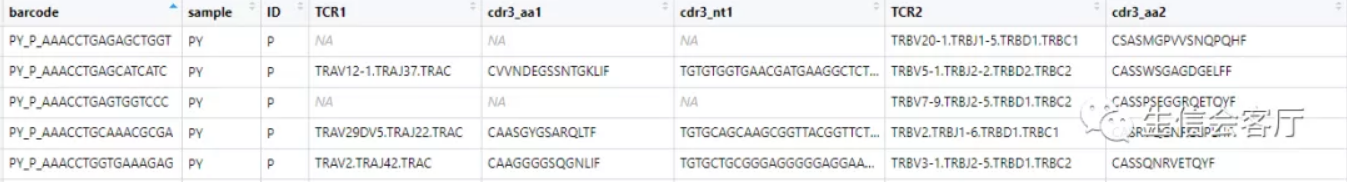
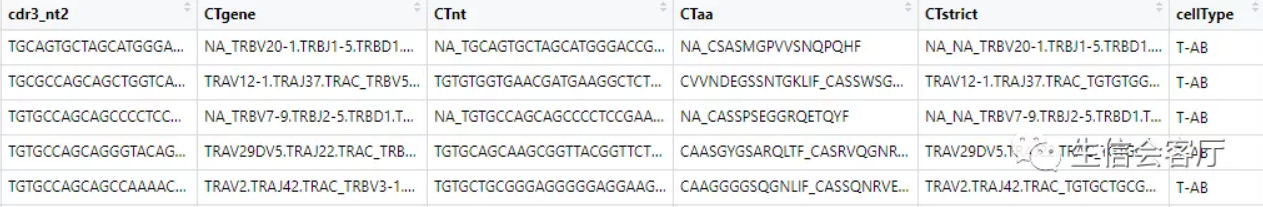
可以看到barcode都加上了由samples和ID组成的前缀,并且把每个细胞配对的两条链放在一行,之后的分析都基于配对信息定义的clonotype。下面分析所用的函数,一般都有两个通用参数:
exportTable:默认赋值F,函数分析结果为图形;改为T之后函数分析结果输出为表格。cloneCall:可选"aa", "nt", "gene", "gene+nt",代表用CDR3氨基酸序列、CDR3核苷酸序列、VDJ基因还是VDJ基因+CDR3核苷酸序列中的哪一种来定义克隆型。以下分析主要用"aa"定义clonotype,大家可以根据自己的需要选择其他类型。
##展示每个样本的克隆型数量
p1 <- quantContig(combined, cloneCall="gene+nt", scale = F)
p2 <- quantContig(combined, cloneCall="gene+nt", scale = T)
plotc = p1 + p2
ggsave('VDJ/quantContig.png', plotc, width = 8, height = 4)
#设置exportTable = T,则输出表格而非图形
quantContig(combined, cloneCall="gene+nt", scale = T, exportTable = T
# contigs values total scaled
# 1 2692 PY_P 3208 83.91521
# 2 1513 PY_T 3119 48.50914
# 3 823 PX_P 1068 77.05993
# 4 928 PX_T 1678 55.30393
# 5 1147 PZ_P 1434 79.98605
# 6 764 PZ_T 2768 27.60116
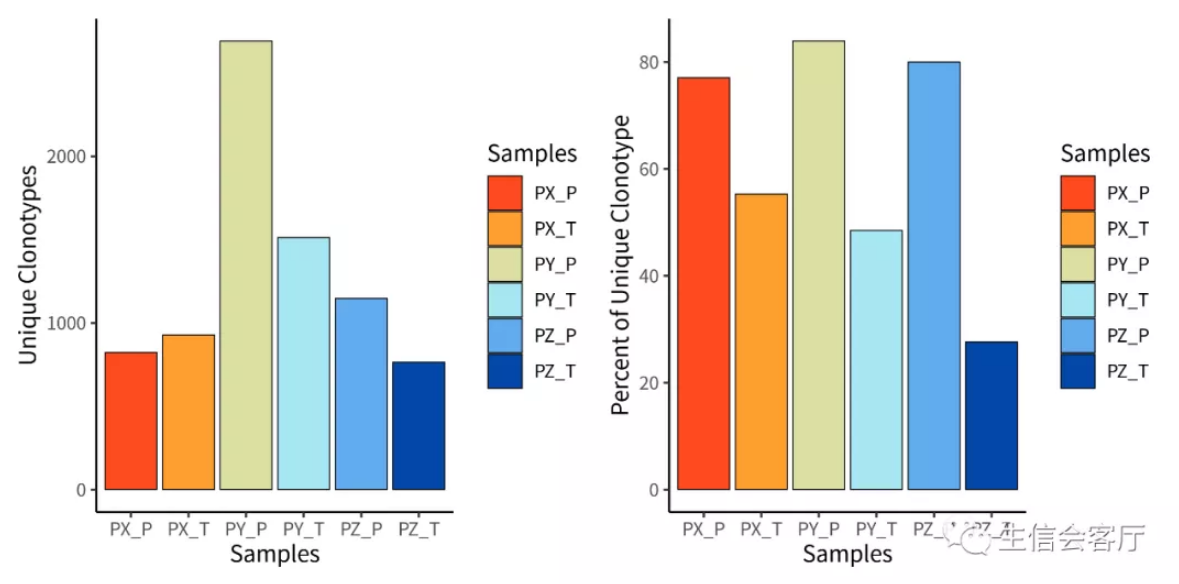 左图展示的是每个样本克隆型总数,右图展示的是克隆型总数/克隆种数
左图展示的是每个样本克隆型总数,右图展示的是克隆型总数/克隆种数
##CDR3序列的长度分布,“aa”代表统计氨基酸序列长度
p1 <- lengthContig(combined, cloneCall="aa", chains = "combined")
p2 <- lengthContig(combined, cloneCall="aa", chains = "single")
plotc = p1 + p2
ggsave('VDJ/lengthContig.png', plotc, width = 8, height = 4)
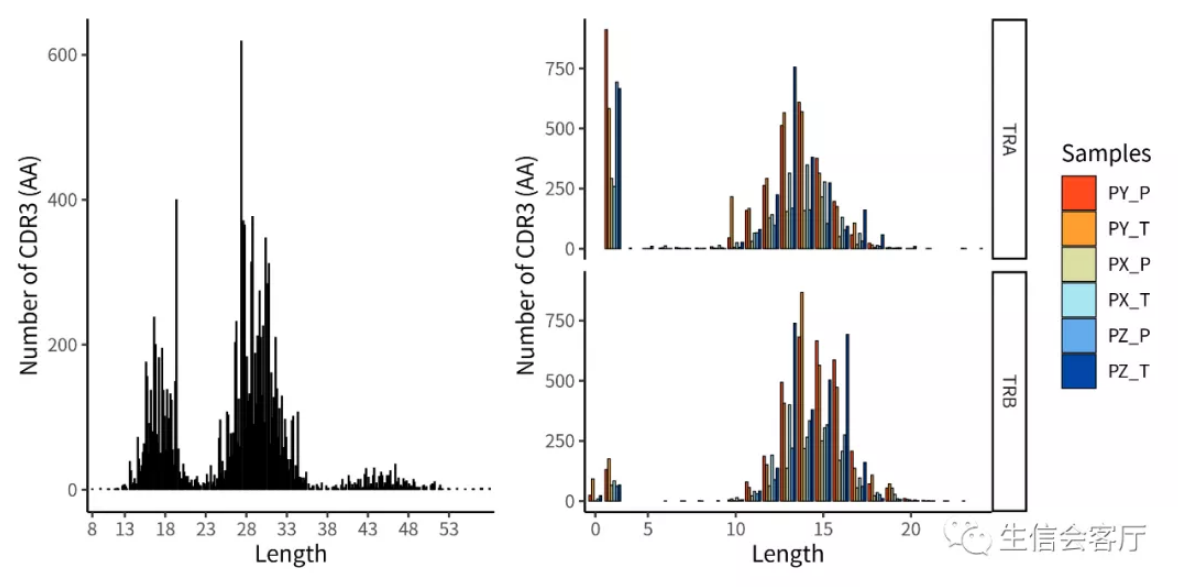
左图展示的是α和β链一起统计,右图展示的是α和β链分开统计
##对比两个样本的克隆型
p1 = compareClonotypes(combined, numbers = 10, samples = c("PX_P", "PX_T"),
cloneCall="aa", graph = "alluvial")
ggsave('VDJ/compareClonotypes.png', p1, width = 8, height = 4)
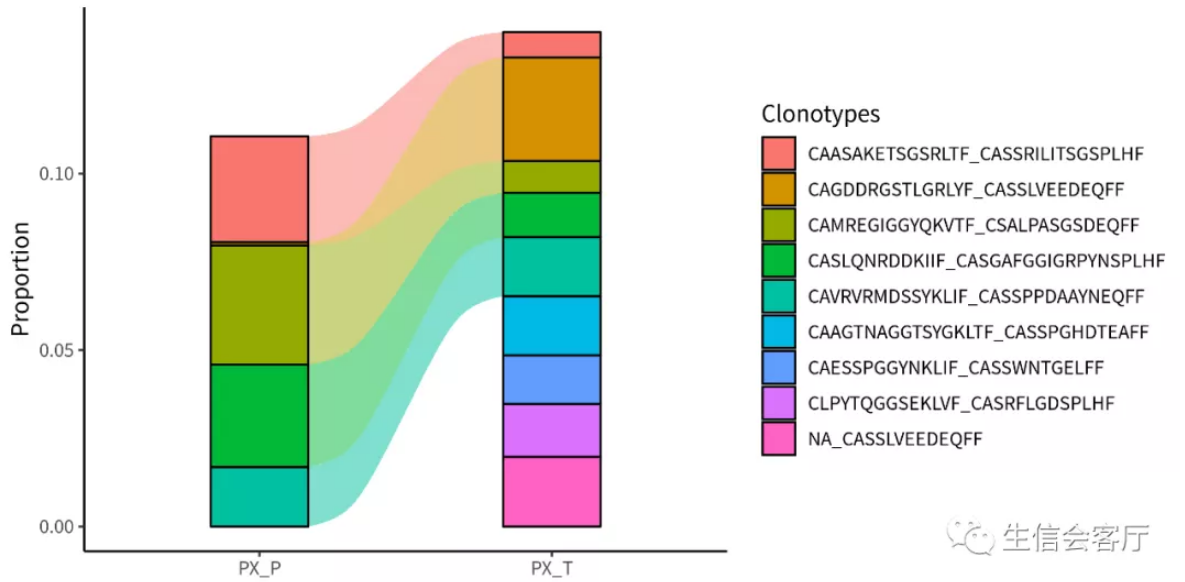
展示排名前10的克隆型在两个样本间的对比情况
##Clonal Space Homeostasis,这个不知道怎么翻译
clonalHomeostasis(combined, cloneCall = "aa")
#此分析将克隆型按其相对丰度分为rare, small, medium, large, hyperexpanded
#5大类,并统计各个类别的占比
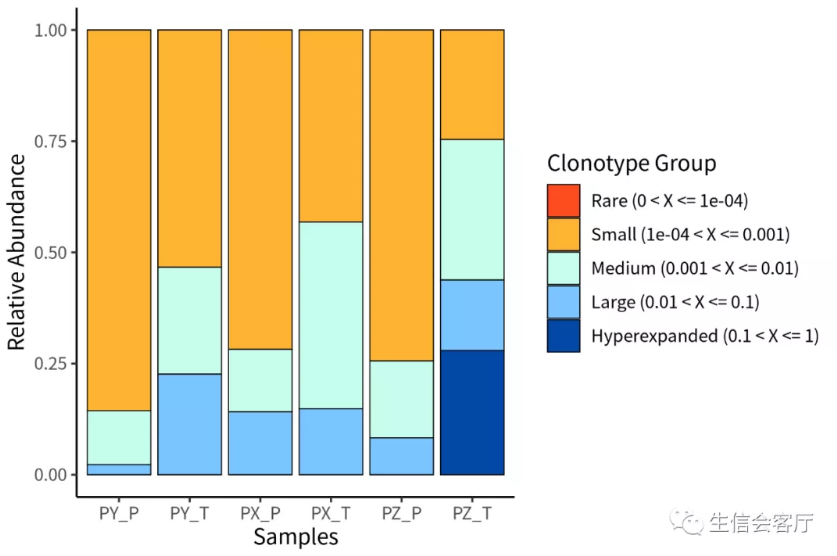
##克隆型分类占比,与上一个分析相似,只是分类方法调整了
clonalProportion(combined, cloneCall = "aa")
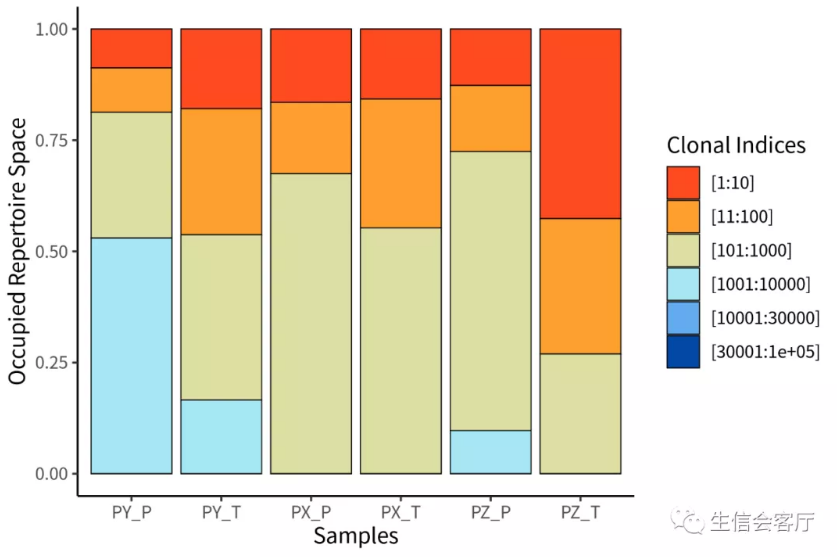
##Overlap Analysis,分析样本相似性
clonalOverlap(combined, cloneCall = "aa", method = "morisita")
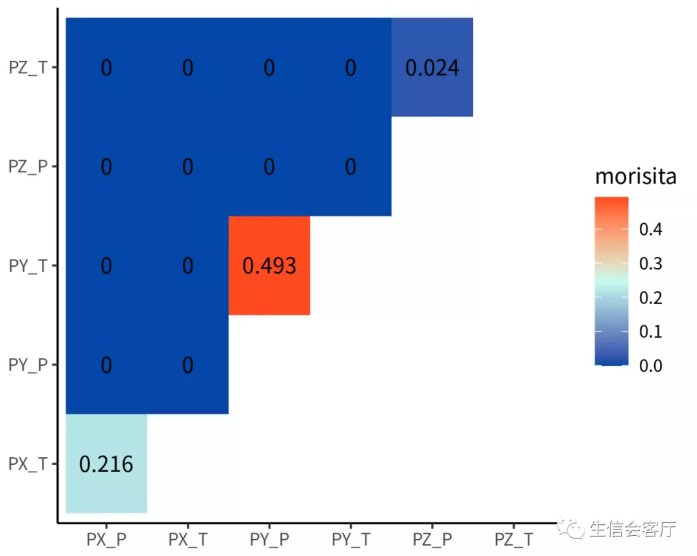
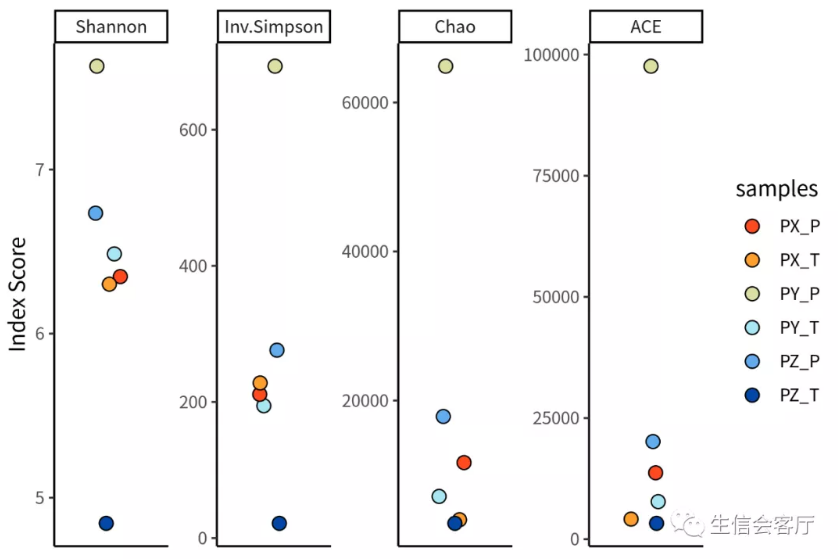
##克隆多样性指数
clonalDiversity(combined, cloneCall = "aa", group = "samples")