单细胞RNA-seq分析介绍
为什么要学习single-cell RNA-seq
在整个人体组织中,细胞类型、状态和相互作用是非常多种多样的,为了更好的了解这些组织和存在的细胞类型,我们需要更高分辨率的技术,而scRNA-seq提供了在单个细胞水平上表达哪些基因的信息,恰好能满足我们的需求。
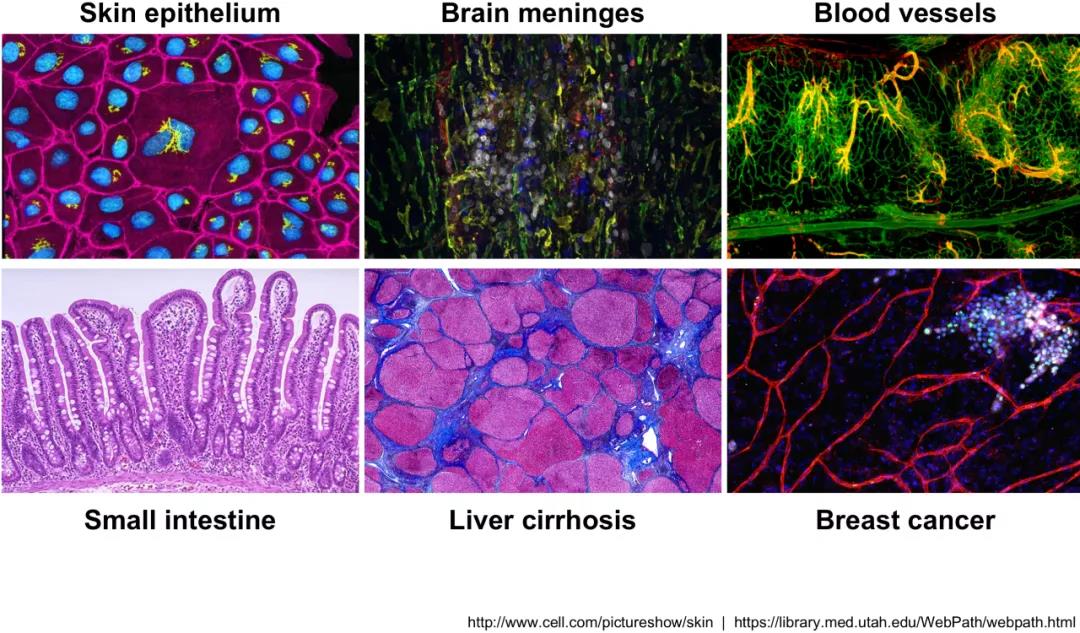
这种令人激动的新方法可以用于:
- 探索组织中存在哪些细胞类型
- 识别未知/稀有的细胞类型或状态
- 阐明分化过程中或跨时间或不同状态下的基因表达变化
- 识别在特定条件下(例如治疗或疾病)在特定细胞类型中差异表达的基因
- 纳入空间,调控和蛋白表达信息,探索细胞类型之间表达的变化
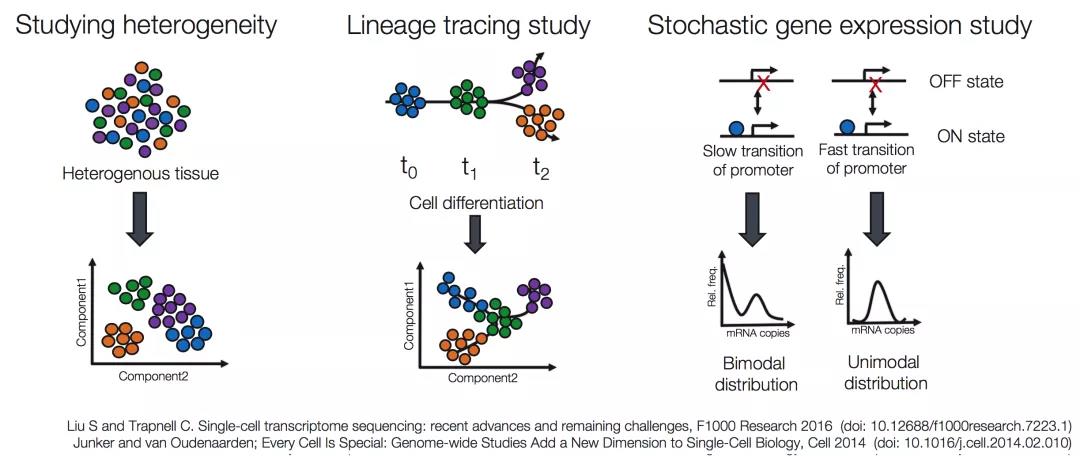
scRNA-seq是解决一些较常见研究的流行方法,比如:
- 细胞异质性研究
- 谱系追踪研究
- 随机基因表达研究
scRNA-seq分析面临的挑战
在scRNA-seq之前,使用大量RNA-seq进行转录组分析,这是一种比较细胞表达平均值的简单方法。如果寻找疾病生物标志物,或者不期望或不关心样品中的大量细胞异质性,则这可能是最佳方法。
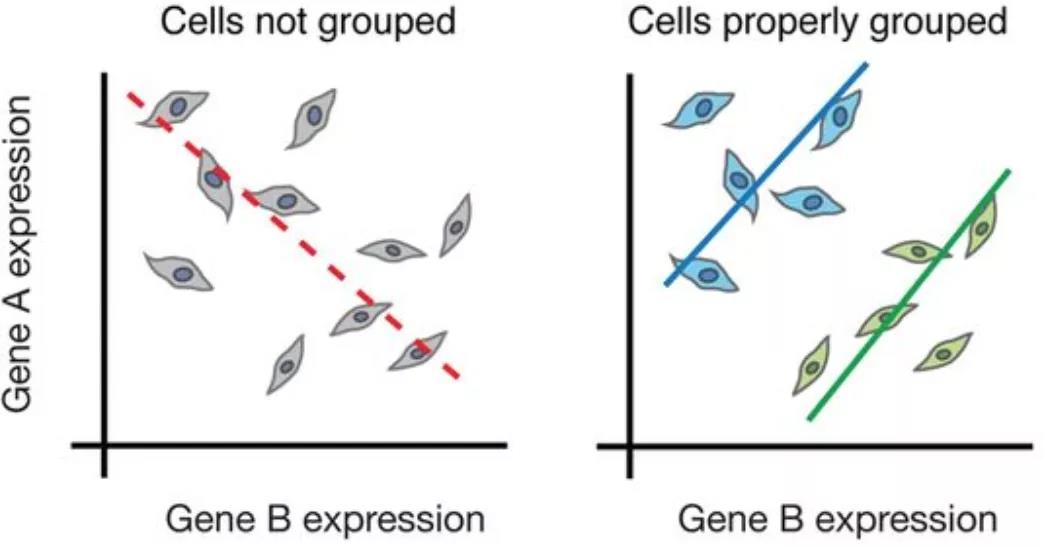
尽管大量RNA-seq可以探索不同条件(例如治疗或疾病)之间基因表达的差异,但无法充分捕获细胞水平的差异。例如,在下面的图像中,如果进行大量分析(左),我们将无法检测到基因A和基因B的表达之间的正确关联。但是,如果我们按细胞类型或细胞状态正确地对细胞进行分组,我们可以看到基因之间的正确相关性。
正如人无完人,技术也一样。尽管scRNA-seq能够在细胞水平上捕获表达,具有诸多优势,但样品的产生和文库的制备更加昂贵,并且分析更为复杂且难以解释,是研究人员不得不面临的挑战。
scRNA-seq数据分析的复杂性:
数据量大
来自scRNA-seq实验的表达数据代表了成千上万个细胞的十万或十万个读数。数据输出要大得多,需要更多的内存进行分析,更大的存储要求以及更多的时间来运行分析。
每个细胞的测序深度很低
对于基于液滴的scRNA-seq方法,测序深度较浅,通常每个细胞仅检测10-50%的转录组。这导致细胞中许多基因的计数为零。但是,在特定的细胞中,基因的零计数可能意味着该基因没有被表达或者是没有检测到转录本。在整个细胞中,具有较高表达水平的基因倾向于具有较少的零。由于这一特性,许多基因在任何细胞中都不会被检测到,细胞间的基因表达也会有很大差异。
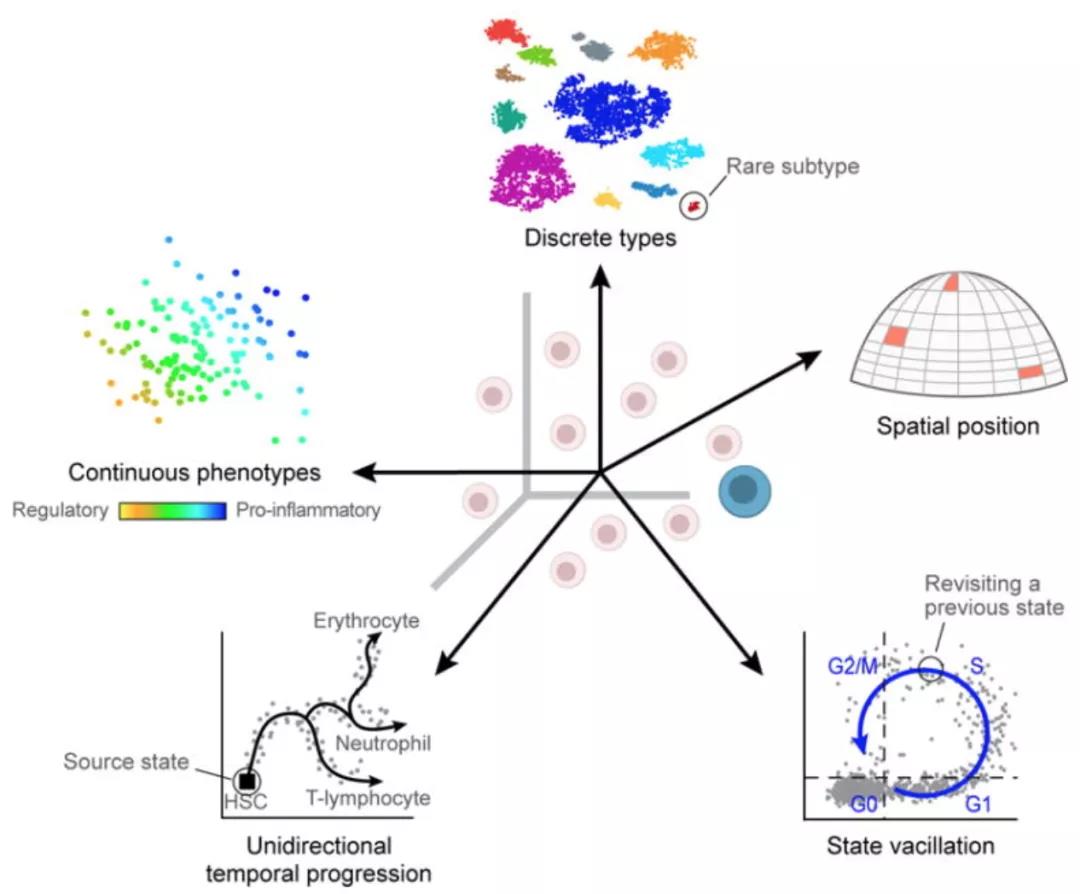
跨细胞/样品的生物学差异
生物学差异的无用来源可能导致细胞之间的基因表达比实际生物细胞类型/状态更相似/不同,这可能会掩盖细胞类型的标识。生物学差异的无用来源(除非实验研究的一部分)包括:
- 转录爆发:并非所有时间都一直打开基因转录。收获时间将决定基因在每个细胞中是打开还是关闭
- RNA处理的速率各不相同:不同的RNA的处理速率不同
- 连续或离散的细胞特性(例如,每个单个T细胞的促炎症潜能):连续的表型是基因表达中定义明确的变量,有时很难将连续的与离散的分离
- 环境刺激:细胞的局部环境可以根据空间位置,信号分子等影响基因的表达
- 时序变化:基本的细胞流动过程,例如细胞周期,会影响单个细胞的基因表达谱。
跨细胞/样品的技术差异
技术差异来源可能导致细胞间的基因表达因技术差异而变的更加相似或不同,而不是生物细胞的类型/状态变化造成的,这会模糊细胞类型的一致性。
技术差异的来源包括:
- 细胞特异性捕获效率:不同细胞捕获的转录物数量不同,导致测序深度不同(例如,转录组的10-50%)。
- 文库质量:降解的RNA,低存活力/濒死细胞,大量自由漂浮的RNA,离解差的细胞以及细胞定量不准确可能导致质量指标降低
- 扩增偏差:在文库制备的扩增步骤中,并非所有转录本都扩增到相同水平。
- 批处理效应:对于scRNA-Seq分析,批处理效应是一个重要的问题,因为您可以仅由于批处理效应就看到表达上的显着差异。
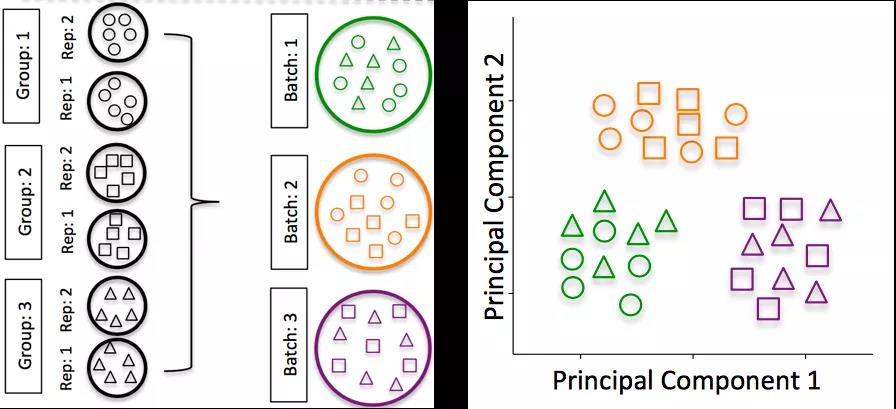
如何知道是否有批次效应
- 是否在同一天进行了所有RNA的分离?
- 是否在同一天进行了所有文库的准备工作?
- 是否由同一个人对所有样品进行RNA分离/文库制备?
- 是否对所有样品使用相同的试剂?
- 是否在同一地点进行RNA分离/文库制备?
- 如果答案为“否”,那么你就有批次效应
关于批次效应的最佳做法:
- 如果可能,尽量以避免批次效应的方式设计实验
- 如果无法避免批量处理
- 不要分批混淆实验
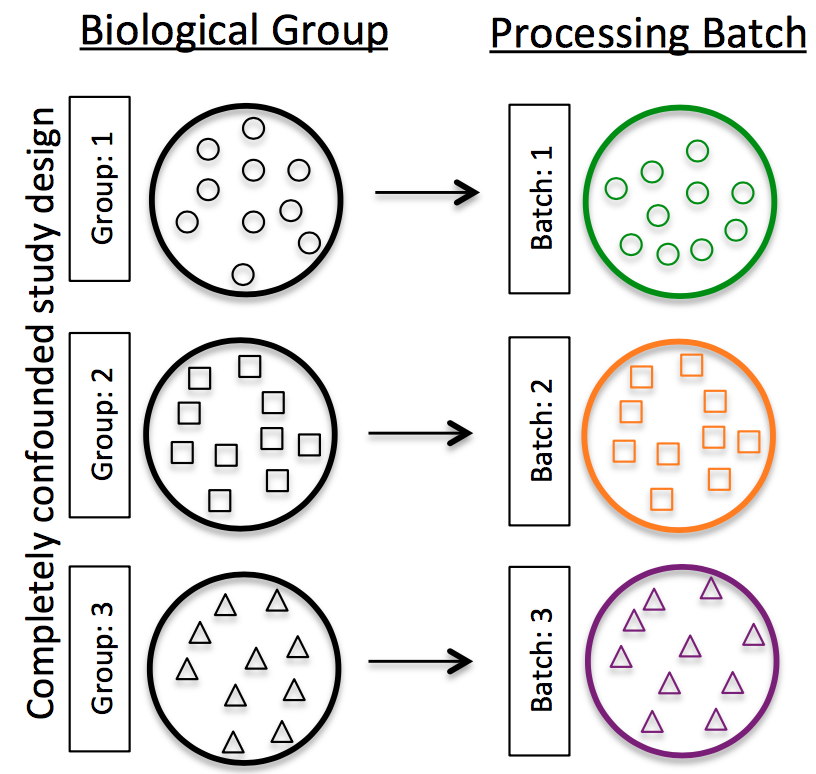
不要对不同的样本组进行分批重复,跨批次拆分不同样本组的重复样本。如果跨条件进行DE或在总体水平上得出结论,则重复越多越好(肯定大于2)。如果使用一次准备一个库的inDrops,则交替使用样品组(例如,不要先准备所有对照库,然后准备所有处理库)
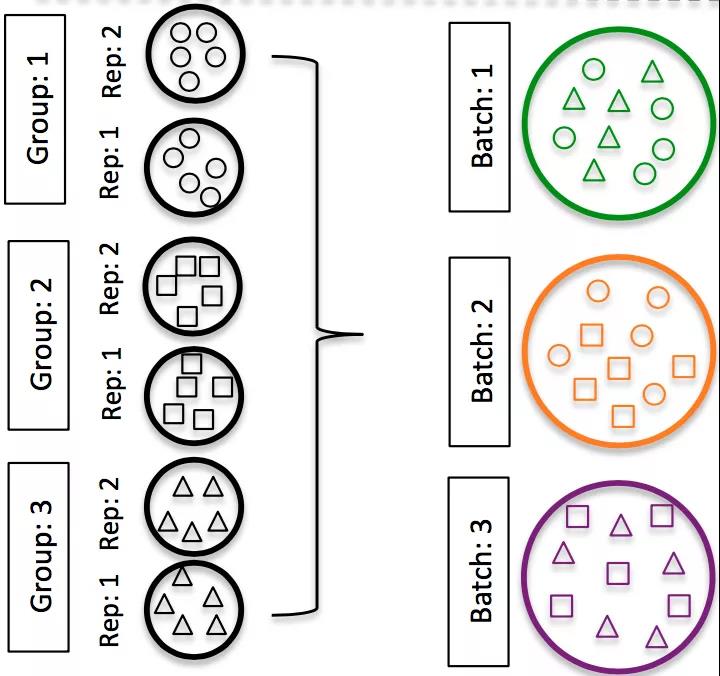
不要在您的实验性元数据中包含批处理信息。在分析过程中,我们可以消除由于批次引起的差异,也可以跨批次进行整合,因此,只要我们掌握了这些信息,就不会影响我们的结果。
结论
虽然scRNA-seq是一种功能强大且有见地的可以从单细胞水平分析基因表达的方法,但仍存在许多挑战和变异来源,这可能会使数据分析变得复杂或有限。
总体而言,我们建议以下内容:
- 除非对感兴趣的实验问题有必要,否则不要进行单细胞RNA-seq。您首先要思考,您是否能使用批量测序来解决你的问题吗?这更简单且成本更低?也许FACS可以对样品进行分类,以便进行批量分析?
了解您要解决的实验性问题的详细信息。以便根据具体实验来确定文库制备方法和分析工作流程
尽可能避免使用技术上的差异来源
- 在实验开始之前与专家讨论实验设计
- 同时从样品中分离RNA
- 同时准备样品库或备用样品组,以避免批次混淆
- 不要混淆性别,年龄或批次的样本组